

 Sequencer Products: SEQ ALL
Sequencer Products: SEQ ALL












 Technologies
Technologies Applications
Applications Online Resources
Online Resources Data Bulletins
Data Bulletins Service & Support
Service & Support Introduction
Introduction Newsroom
Newsroom Doing Business With Us
Doing Business With Us Creative Club
Creative Club




The nucleic acid detection technology based on real-time fluorescent RT-PCR method has played an important role in the rapid identification and diagnosis of coronaviruses. However, to study the origin of coronaviruses, their evolution and pathogenic mechanisms, it is necessary to obtain complete viral genome information, which requires high-throughput sequencing and viral sequence assembly.
In order to fully reveal the relevant features of the new coronavirus, MGI can provide integrated packages for high-throughput sequencing, sequence assembly, and phylogenetic analysis of new coronaviruses, and has assisted Centers for Disease Control and Prevention (CDC) across the country to successfully assemble full-length sequences of the coronavirus. The results show that they are highly consistent with published reference genome sequences.
Difficulties and requirements in the assembly process of Novel Coronavirus
As everyone knows, high-throughput sequencing can complement the RT-PCR method in the identification and diagnosis of new coronaviruses, which can not only improve the positive detection rate, but also identify co-infection pathogens and provide more information about the pathogens of possible infections. Importantly, it can also assemble virus sequences to obtain the full-length genome information of the virus, providing a research basis for tracing the source of the virus, monitoring the trend of virus mutations, and exploring the pathogenic mechanism.
In order to obtain the complete viral genome sequence, the widely-used high-throughput sequencing technology is to break the nucleic acid sequence into short fragments for sequencing, and then use the analysis software to assemble short sequences. However, as this is a novel virus, there is no standard experimental protocol with recommended sequencing depth, sequencing accuracy and duplication rate. Thus, there are several difficulties to overcome in order to assemble the genome sequence from a large number of short sequences:
First, sequencing errors will inevitably lead to low accuracy in certain regions. Second, incomplete coverage of genomic sequences and high sequencing duplication rates will affect the accuracy and integrity of assembly. Finally, in metatranscriptome sequencing samples, the human-derived sequences account for more than 85%, and the pathogenic sequences account for only about 5%, which makes the assembly of viral genome sequences more difficult.
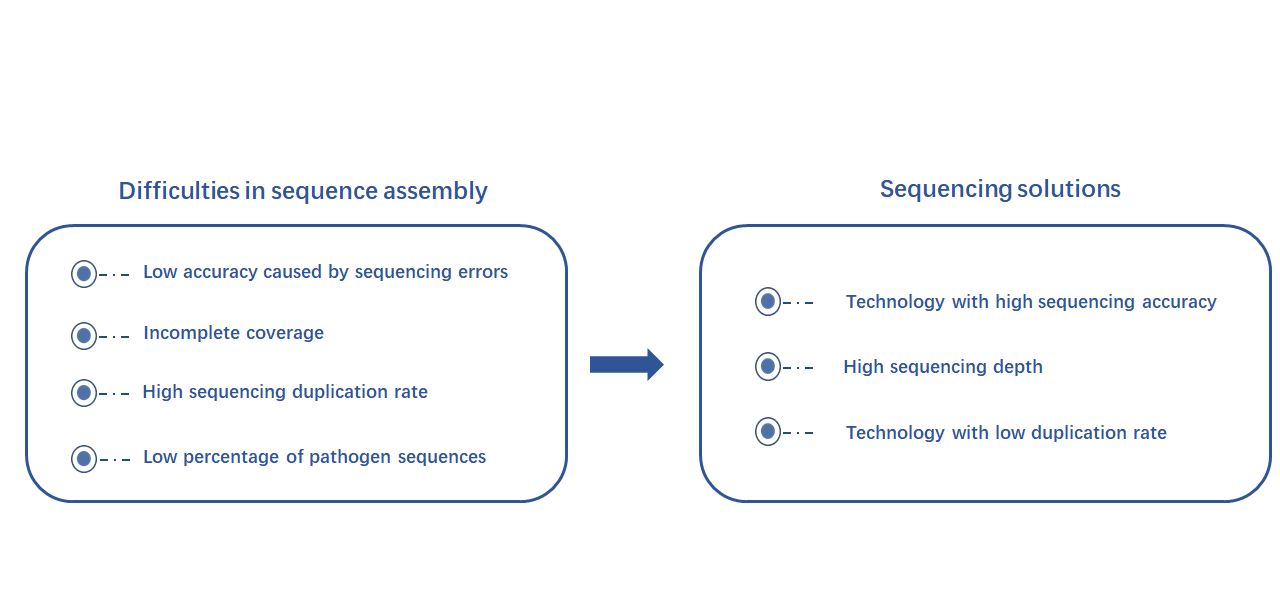
Figure 1 Difficulties in sequence assembly and its requirements for sequencing packages
Optimize sequencing strategy to ensure virus sequence information integrity
In order to solve the problems encountered in the assembly process of the novel coronavirus sequences described above, MGI can provide complete packages including library construction, high-throughput sequencing, sequence assembly, and phylogenetic analysis.
In the process of library construction, in order to avoid large differences in the content of extracted nucleic acids due to uncertainty during sampling, storage and transportation, MGI suggests two packages: for samples with high amount of nucleic acid , it is suggested to do rRNA depletion before library construction to increase the percentage of viral sequences. For samples with less amount of nucleic acid , it is recommended to prepare RNA library directly without rRNA depletion to reduce nucleic acid loss, increase the success rate of library construction, and increase the depth of sequencing.
Secondly, in the sequencing process, MGISEQ-200 (DNBSEQ-G50) sequencer is recommended, because it is not only small and flexible, but also efficient. It has assisted Centers for Disease Control and Prevention across the country to complete identification and successfully assemble the first novel coronavirus cases in various places.
Finally, the pathogenic identification system was used to analyze the data of the new coronavirus sequence and the IDBA method was used to complete the assembly.
In this way, even without removing the host, MGI sequencing package is able to provide enough sequencing data for virus sequence assembly and ensure the integrity of the sequence information.
Centers of Disease Control and Prevention using MGISEQ-200 (DNBSEQ-G50) to decipher coronavirus
Following is an example how one of the CDCs using MGI package to confirm the first suspected cases of novel coronavirus pneumonia and perform virus sequence assembly on respiratory samples:
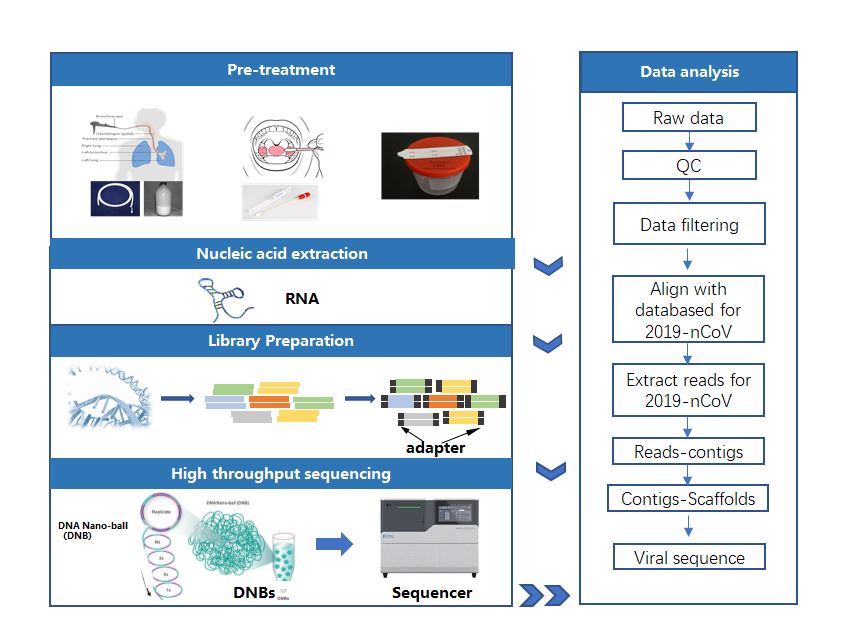
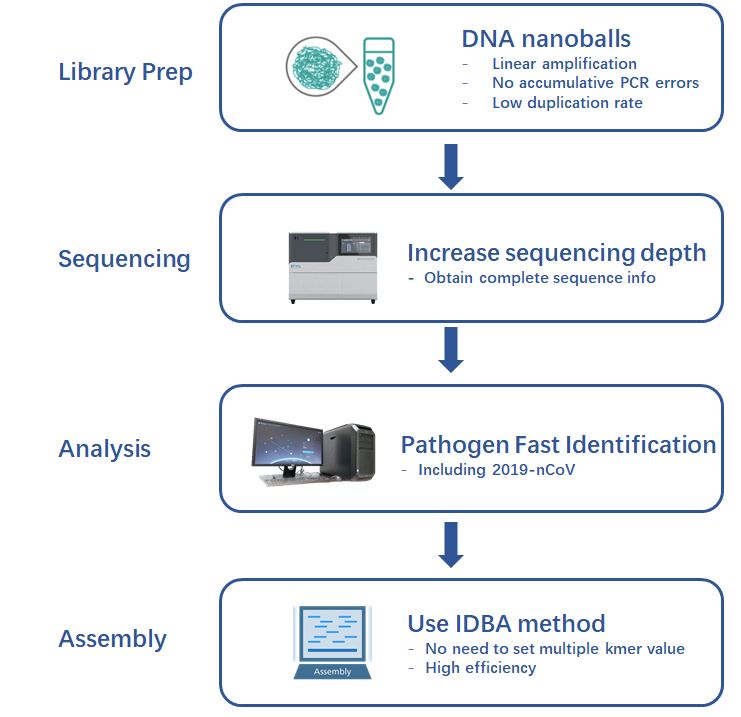
Figure 3 Whole process of obtaining the whole genome sequence of novel coronavirus
The whole process of obtaining the whole genome sequence of novel coronavirus
January 20, Library Preparation
For different nucleic acid samples, the team adopted different library building strategies and used the MGIEasy RNA library preparation reagent kit to construct the library. The libraries were obtained after a series of steps including reverse transcription, adapter ligation, PCR amplification, purification, etc. Then, it uses rolling circle amplification technology to prepare DNA nanoballs.
Figure 4 MGIEasy RNA library preparation reagent kit
January 21, Sequencing
Based on the MGISEQ-200 (DNBSEQ-G50) platform, high-depth sequencing with 300M reads were performed on the respiratory samples of the first case reported in the region.

Figure 5 MGISEQ-200 (DNBSEQ-G50) sequencer run by the CDC
January 22,Data Analysis
Generated 32Gb data with a total of 318M reads. Combined with the Pathogen Fast Identification system, a total of 2,337,442 new coronavirus reads were identified.
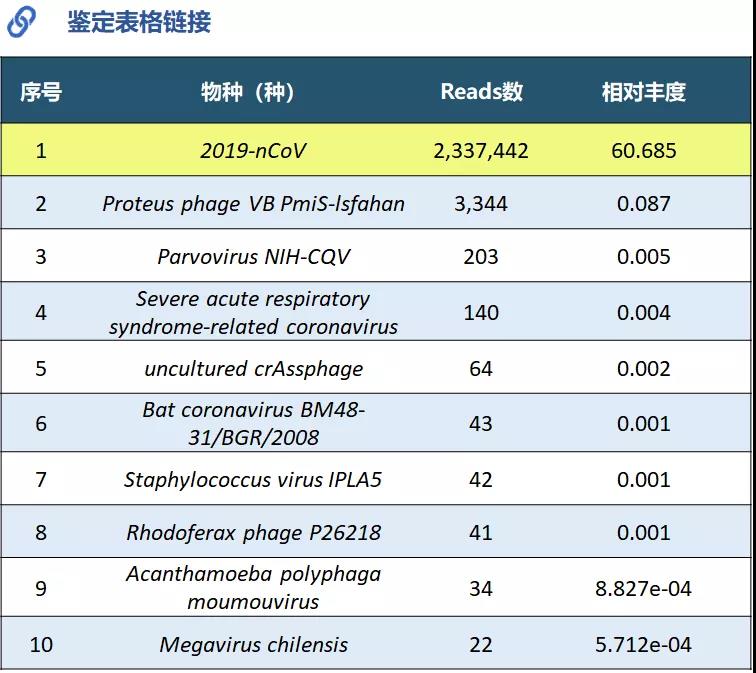
Figure 6 Analysis report with virus identification results
January 22, Sequence Assembly
The analysis software automatically extracted the 2,337,442 new coronavirus reads from all the sequences. The IDBA method with high assembly efficiency was used, and a full-length 29.9kb genome sequence was obtained.
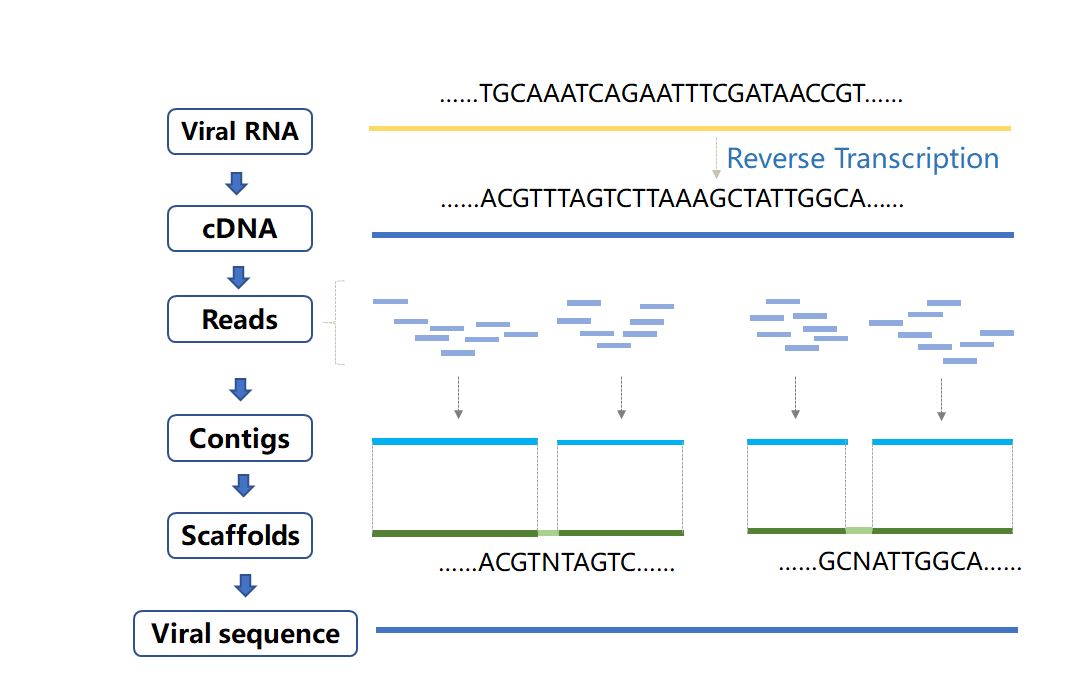
Figure 7 Assembly process of viral genome sequences
Although our knowledge of the novel coronavirus is still limited, obtaining the whole genome sequence of novel coronavirus through metatranscriptome sequencing and viral sequence assembly can help reveal the relevant characteristics of the virus. By comparing the similarity of whole genome sequences and mutation site analysis can provide important reference information for constructing evolutionary trees, tracing virus sources, tracking mutation paths, and understanding the pathogenic mechanism, etc. to help fight the epidemic.








