

 Sequencer Products: SEQ ALL
Sequencer Products: SEQ ALL












 Technologies
Technologies Applications
Applications Online Resources
Online Resources Data Bulletins
Data Bulletins Service & Support
Service & Support Introduction
Introduction Newsroom
Newsroom Doing Business With Us
Doing Business With Us Creative Club
Creative Club




Thanks to high-throughput sequencing technologies, shotgun metagenomic methods were made possible and had effectively transformed microbiology. Today, advances in both short- and long-read technologies are overcoming many of the previous challenges affecting metagenomic profiling, especially of highly complex samples and environment.
Researchers from France's National Research Institute for Agriculture, Food, and Environment (INRAE) examined the performance of seven short- and long-read sequencing platforms in analyzing high-complexity metagenomic samples. The study, published in the Nature Portfolio journal Scientific Data, ran mock samples between 2018 and 2019 on various mainstream sequencers at the time, including MGI's DNBSEQ-T7* and DNBSEQ-G400*.
Within this wide range of sequencing technologies tested, DNBSEQ-T7* was recognized for its ultra-high throughput and excellent accuracy. "We were surprised by the T7’s performance*," said senior author Mathieu Almeida, a research fellow at INRAE. "It provides ultra-deep sequencing in a single run with similar low error rate compared to the other platforms, making it at the time of our study one of the most affordable technologies for metagenomic sequencing."
In the study, three uneven synthetic microbial communities were constructed, consisting of up to 87 genomic microbial strains DNAs each and spanning 29 bacterial and archaeal phyla. They represented some of the most complex and diverse communities used for sequencing technology comparisons. The mock1 (71 strains) was sequenced using all platforms, mock2 (64 strains) was additionally sequenced to estimate the impact of various microbial richness, while MGI's platforms were not performed on mock3.
To assess the impact of sequencing depth, the team ran a subsampling analysis and compared observed and theoretical genome abundances across samples at multiple depth from 10,000 to 1 million reads. Overall, Spearman rank correlations for all platforms were high at above 0.9 when mapping at least 100,000 reads. Among them, the correlations of T7* and G400* were the best in mock1 and remained excellent in mock2.
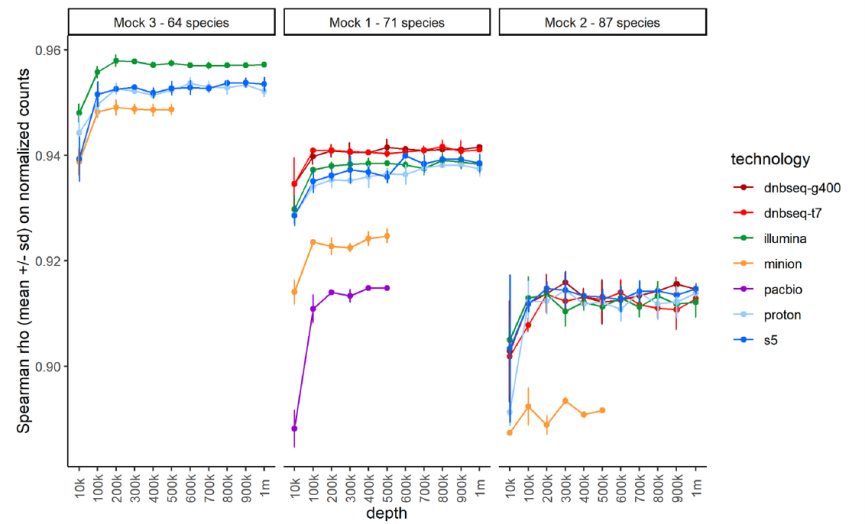
Overall comparison between observed and excepted mock compositions for each platform
In addition, differential analysis between observed and excepted species abundances was performed in mock1. Results showed that over or under abundance estimation for most genomes had little to do with the sequencing platform, read length, taxonomy, GC-content, genome size and genome completeness, even at a low depth of 500,000 reads. In fact, most genomes were accurately estimated on all sequencers, with the observed normalized abundances generated by T7* charting very close to the excepted values.
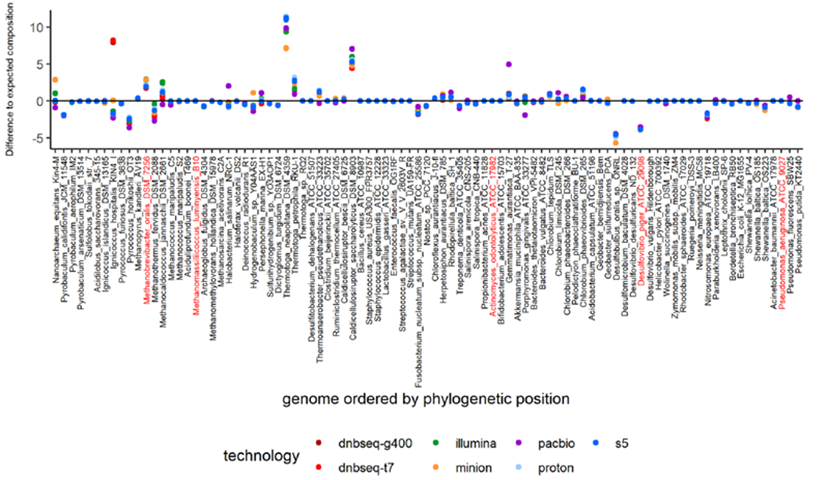
Differential plot between observed and excepted species abundances in mock1
Based on performance analyses of the different sequencers, the study formed a microbial metagenomic sequencing benchmarking database, providing researchers and scientists a comprehensive and authentic reference for sequencing platform selection. In particular, the findings demonstrated the promising value of MGI's DNBSEQ-T7* in metagenomic sequencing.
Boasting high stability and accuracy as shown in the data, combined with outstanding throughput, T7* makes a strong platform for the identification of species and functional genes in highly complex microbial communities. Its upgraded biochemical, fluidics, and optical systems are not only making sequencing more efficient and productive, but also continuing to support research into the structure and diversity of microbial communities.
*Unless otherwise informed, StandardMPS and CoolMPS sequencing reagents, and sequencers for use with such reagents are not available in Germany, Spain, UK, Sweden, Belgium, Italy, Finland, Czech Republic, Switzerland, Portugal, Austria, and Romania. Unless otherwise informed, StandardMPS sequencing reagents, and sequencers for use with such reagents are not available in Hong Kong. No purchase orders for StandardMPS products will be accepted in the USA until after January 1, 2023.








